昇思学习打卡营第30天|在量子化学计算中应用量子变分求解器
dist = 1.5spin = 0geometry,basis,run_scf=1,run_fci=1结果通过本案例,我们了解了量子变分求解器(VQE)在量子化学计算中的应用。VQE利用变分原理和量子计算的优势,能够高效地求解分子体系的基态能量。使用MindSpore Quantum结合经典优化算法,我们成功模拟了LiH分子的基态能量。该方法展示了量子计算在化学模拟中的巨大潜力,为未来更复杂的分
使用场景
量子化学计算在现代科学研究中至关重要,用于模拟和预测分子结构、化学反应和材料性能。然而,传统的量子化学计算方法在处理复杂分子体系时面临巨大挑战。量子变分求解器(VQE)作为一种量子-经典混合算法,能够有效解决这一问题。VQE可以在量子计算机上实现多项式复杂度下对薛定谔方程的高精度求解,为大规模分子体系的模拟提供了可能。
算法原理
变分原理
变分原理用于估计量子系统的基态能量。通过选择一个参数化的试探波函数,并优化这些参数,使得波函数的期望值尽可能接近实际的基态能量。具体步骤如下:
- 制备初态:利用Hartree-Fock方法生成的初态波函数。
- 定义波函数拟设:如UCCSD(幺正耦合簇单双激发)方法。
- 量子线路实现:将波函数拟设转化为参数化的量子线路。
- 优化变分参数:在经典计算机上使用优化算法(如BFGS)更新变分参数。
- 测量与迭代:在量子计算机上测量能量和梯度,不断优化参数,直到收敛。
代码实现
以下代码展示了如何使用MindSpore Quantum实现VQE算法,并求解LiH分子的基态能量。
环境准备
首先,安装所需模块:
pip install mindquantum openfermion openfermionpyscf
导入依赖
from openfermion.chem import MolecularData
from openfermionpyscf import run_pyscf
from mindquantum.core.gates import X
from mindquantum.core.circuit import Circuit
from mindquantum.core.operators import Hamiltonian
from mindquantum.simulator import Simulator
from mindquantum.algorithm.nisq import generate_uccsd
import mindspore as ms
import numpy as np
from scipy.optimize import minimize
定义分子结构
dist = 1.5
geometry = [
["Li", [0.0, 0.0, 0.0 * dist]],
["H", [0.0, 0.0, 1.0 * dist]],
]
basis = "sto3g"
spin = 0
molecule_of = MolecularData(
geometry,
basis,
multiplicity=2 * spin + 1,
data_directory='./'
)
molecule_of = run_pyscf(
molecule_of,
run_scf=1,
run_ccsd=1,
run_fci=1
)
print("Hartree-Fock energy: %20.16f Ha" % (molecule_of.hf_energy))
print("CCSD energy: %20.16f Ha" % (molecule_of.ccsd_energy))
print("FCI energy: %20.16f Ha" % (molecule_of.fci_energy))
molecule_of.save()
molecule_file = molecule_of.filename
print(molecule_file.split('/')[-1])
制备初态和波函数拟设
hartreefock_wfn_circuit = Circuit([X.on(i) for i in range(molecule_of.n_electrons)])
print(hartreefock_wfn_circuit)
ansatz_circuit, init_amplitudes, ansatz_parameter_names, hamiltonian_QubitOp, n_qubits, n_electrons = generate_uccsd(molecule_file, threshold=-1)
total_circuit = hartreefock_wfn_circuit + ansatz_circuit
total_circuit.summary()
print("Number of parameters: %d" % (len(ansatz_parameter_names)))
定义优化函数和VQE流程
sim = Simulator('mqvector', total_circuit.n_qubits)
molecule_pqc = sim.get_expectation_with_grad(Hamiltonian(hamiltonian_QubitOp), total_circuit)
n_params = len(total_circuit.params_name)
p0 = np.zeros(n_params)
f, g = molecule_pqc(p0)
print("Energy: ", f, "\nshape: ", f.shape, '\n')
print("Gradient: ", g, "\nshape: ", g.shape)
def fun(p0, molecule_pqc, energy_list=None):
f, g = molecule_pqc(p0)
f = np.real(f)[0, 0]
g = np.real(g)[0, 0]
if energy_list is not None:
energy_list.append(f)
if len(energy_list) % 5 == 0:
print(f"Step: {len(energy_list)},\tenergy: {f}")
return f, g
energy_list = []
res = minimize(fun, p0, args=(molecule_pqc, energy_list), method='bfgs', jac=True)
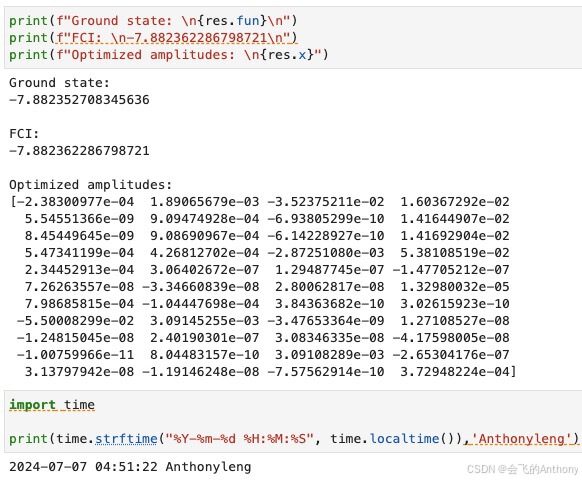
print(f"Ground state: \n{res.fun}\n")
print(f"FCI: \n-7.882362286798721\n")
print(f"Optimized amplitudes: \n{res.x}")
结果
学习总结
通过本案例,我们了解了量子变分求解器(VQE)在量子化学计算中的应用。VQE利用变分原理和量子计算的优势,能够高效地求解分子体系的基态能量。使用MindSpore Quantum结合经典优化算法,我们成功模拟了LiH分子的基态能量。该方法展示了量子计算在化学模拟中的巨大潜力,为未来更复杂的分子体系计算提供了可行的途径。通过本教程,我们学会了如何构建量子线路、定义波函数拟设以及使用经典优化器进行参数优化,从而完成了VQE的整个流程。
如果你觉得这篇博文对你有帮助,请点赞、收藏、关注我,并且可以打赏支持我!
欢迎关注我的后续博文,我将分享更多关于人工智能、自然语言处理和计算机视觉的精彩内容。
谢谢大家的支持!

昇腾计算产业是基于昇腾系列(HUAWEI Ascend)处理器和基础软件构建的全栈 AI计算基础设施、行业应用及服务,https://devpress.csdn.net/organization/setting/general/146749包括昇腾系列处理器、系列硬件、CANN、AI计算框架、应用使能、开发工具链、管理运维工具、行业应用及服务等全产业链
更多推荐

 5
5 0
0- 0



 已为社区贡献30条内容
已为社区贡献30条内容

所有评论(0)