开源scRNA Tools 1. |代码分享:使用 ascend R 包从质控到差异表达全流程解析单细胞数据
ascend(Analysis of Single Cell Expression, Normalisation and Differential expression) 是一个基于R 语言开发的综合性工具包,专为单细胞 RNA 测序 (scRNA-seq)数据分析而设计。该包旨在提供用户友好、快速且稳健的分析流程,涵盖了从原始数据处理到下游生物学解读的各个阶段。ascend构建于类之上,利用核心
介绍
ascend (Analysis of Single Cell Expression, Normalisation and Differential expression) 是一个基于 R 语言开发的综合性工具包,专为单细胞 RNA 测序 (scRNA-seq) 数据分析而设计。该包旨在提供用户友好、快速且稳健的分析流程,涵盖了从原始数据处理到下游生物学解读的各个阶段。ascend 构建于 SingleCellExperiment 类之上,利用核心的 EMSet (Expression and Metadata Set) 对象来统一管理表达数据与元数据。其主要功能包括数据质量控制 (QC)、标准化、降维(如 PCA)、聚类以及差异表达分析。此外,该包支持通过 BiocParallel 进行多核并行计算以提升处理效率,并封装了 scran、DESeq 等第三方流行工具,适用于分析来自 10x Chromium、DropSeq 和 inDrop 等多种平台的单细胞转录组数据。

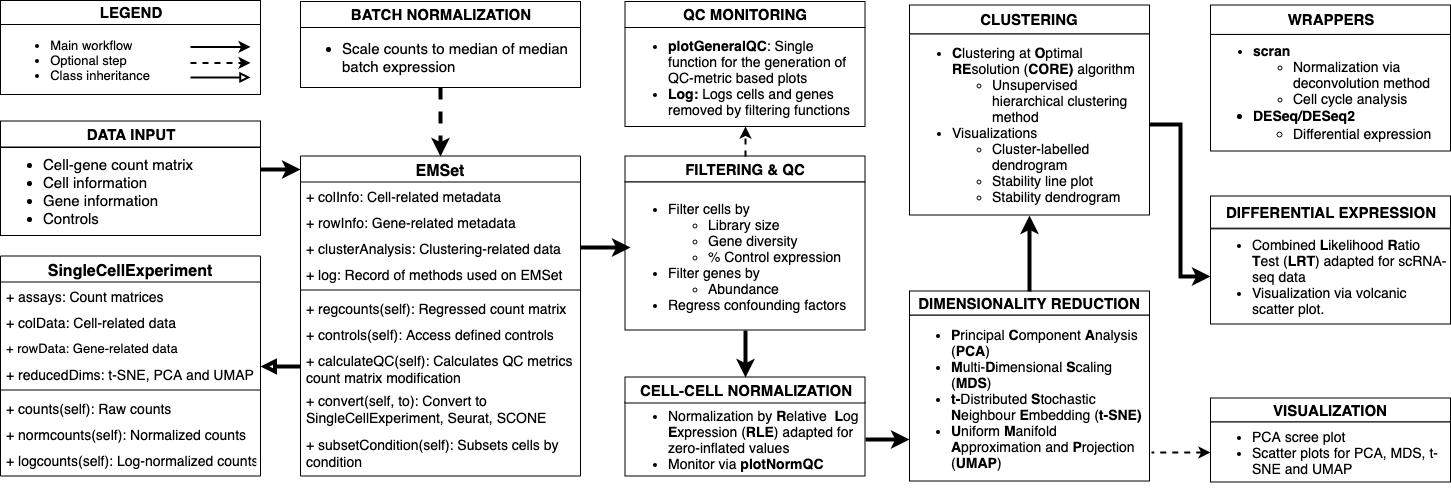
- Link:https://github.com/powellgenomicslab/ascend
适用条件
- 运行环境:依赖 R 语言环境(版本需 >= 3.5.0,推荐 3.6+),且需要在安装前配置好 Rcpp 和 RcppArmadillo 编译环境。
- 数据类型:适用于基于读取计数 (Read counts) 或 UMI 计数的基因-细胞表达矩阵,支持 Chromium、DropSeq 和 inDrop 等平台数据。
- 计算资源:支持 Windows、Linux 和 MacOS 系统,建议在多核硬件上配置 BiocParallel 以实现并行计算加速。
输入数据与功能输出
| 类型 | 描述 |
|---|---|
| 输入数据 | 1. 表达矩阵:行代表基因/转录本,列代表细胞的计数矩阵(Read 或 UMI counts)。 2. 元数据:包含细胞(如批次、样本信息)和基因的相关注释信息。 3. EMSet 对象:上述数据被加载到 ascend 的核心容器对象中进行存储和操作。 |
| 功能输出 | 1. 质控结果:识别并过滤低质量细胞/基因的指标与图表。 2. 标准化矩阵:消除技术噪音后的数据(支持封装 scran 方法)。3. 降维坐标:PCA、t-SNE 等降维分析结果,用于数据探索。 4. 聚类分群:将细胞划分为不同的亚群 (Clusters)。 5. 差异分析:识别不同亚群间的标记基因 (Marker Genes) 及差异表达结果。 |
Demo
# 1. 加载包和数据
library(ascend)
library(ggplot2) # 用于绘图
# 加载示例原始数据
data(raw_set)
EMSet <- raw_set
# 查看 Count 矩阵前几行
counts <- counts(EMSet)
print(counts[1:5, 1:5])
# -----------------------------------------------------------
# 2. 管理细胞元数据 (colInfo)
# -----------------------------------------------------------
col_info <- colInfo(EMSet)
# 修改批次信息:将 Batch 1/2 替换为 THY1 Positive/Negative
batch_list <- col_info$batch
batch_list[which(batch_list == 1)] <- "Positive"
batch_list[which(batch_list == 2)] <- "Negative"
# 将其添加为新的一列并更新对象
col_info$THY1 <- batch_list
colInfo(EMSet) <- col_info
print(colInfo(EMSet))
# 基于特定基因表达标记细胞
gene_markers <- c("POU4F1", "POU4F2", "POU4F3")
gene_markers <- gene_markers[which(gene_markers %in% rownames(EMSet))]
EMSet <- addGeneLabel(EMSet, gene = gene_markers)
print(colInfo(EMSet))
# -----------------------------------------------------------
# 3. 管理基因信息与 ID 转换 (rowInfo)
# -----------------------------------------------------------
row_info <- rowInfo(EMSet)
# 演示:创建一个转换为 ENSEMBL ID 的新对象
ensembl_set <- convertGeneID(EMSet, new.annotation = "ensembl_gene_id")
print(rowInfo(ensembl_set))
print(counts(ensembl_set)[1:5, 1:5])
# -----------------------------------------------------------
# 4. 设置与移除对照基因 (Controls)
# -----------------------------------------------------------
# 定义线粒体(Mt)和核糖体(Rb)基因
control_list <- list(Mt = grep("^Mt-", rownames(EMSet), ignore.case = TRUE, value = TRUE),
Rb = grep("^Rps|^Rpl", rownames(EMSet), ignore.case = TRUE, value = TRUE))
# 设置对照
controls(EMSet) <- control_list
print(controls(EMSet))
# 演示:创建一个移除对照基因的新对象
NoControl_EMSet <- excludeControl(EMSet, control = c("Mt", "Rb"))
# -----------------------------------------------------------
# 5. 对象操作与数据获取
# (注意:此处 Demo 切换到了一个已分析好的完整数据集 'analyzed_set' 以进行后续演示)
# -----------------------------------------------------------
data(analyzed_set)
EMSet <- analyzed_set
# 获取各类矩阵
count_matrix <- counts(EMSet)
norm_matrix <- normcounts(EMSet)
logcounts_matrix <- logcounts(EMSet)
# 获取降维数据
tsne_matrix <- reducedDim(EMSet, "TSNE")
pca_matrix <- reducedDim(EMSet, "PCA")
umap_matrix <- reducedDim(EMSet, "UMAP")
# 像操作数据框一样切片对象 (取前10个细胞和基因)
tiny_EMSet <- EMSet[1:10, 1:10]
# 基于条件取子集 (例如只取 Batch 1)
Batch1_EMSet <- subsetCondition(EMSet, by = "batch", conditions = list(batch = c(1)))
# -----------------------------------------------------------
# 6. 降维与可视化 (t-SNE / UMAP)
# -----------------------------------------------------------
# 运行 t-SNE (使用 PCA 加速)
EMSet <- runTSNE(EMSet, dims = 2, PCA = TRUE, seed = 1, perplexity = 10)
# 查看生成的矩阵
print(reducedDim(EMSet, "TSNE")[1:10, ])
# --- 绘制 t-SNE ---
tsne_plot <- plotTSNE(EMSet, group = "cluster")
# 自定义颜色和主题 (基于 ggplot2)
tsne_plot <- tsne_plot +
scale_color_manual(values=c("#bb5f4c", "#8e5db0", "#729b57")) +
ggtitle("Clusters", subtitle = "tSNE plot") +
theme(legend.position = "bottom")
print(tsne_plot)
# --- 绘制 UMAP ---
umap_plot <- plotUMAP(EMSet, group = "cluster", Dim1 = 1, Dim2 = 2)
# 自定义颜色和主题
umap_plot <- umap_plot +
scale_color_manual(values=c("#bb5f4c", "#8e5db0", "#729b57")) +
ggtitle("Clusters", subtitle = "UMAP plot") +
theme(legend.position = "bottom")
print(umap_plot)
# -----------------------------------------------------------
# 7. 与其他包的转换
# -----------------------------------------------------------
# 转换为 SingleCellExperiment 对象
sce_obj <- convert(EMSet, to = "sce")
print(head(sce_obj))
# 转回 EMSet
EMSet_back <- convert(sce_obj, to = "EMSet")
print(head(EMSet_back))
- 简易生信分析问题可免费解答,复杂问题付费咨询;欢迎投稿需要复现的文献图表,团队可整理和分享一份案例交流学习。
- 期待学术合作者加入团队,磨合后有数据有课题有契机的可合作冲子刊和正刊,另外筹备SCI期刊编委会。
- 承接单细胞空转真核转录组等多组学测序服务(寻因平台) ,欢迎各大医院或课题组咨询,关注6个月以上给予绝对低于市场价的粉丝价。
- 欢迎扩列交流学习聊职业规划等,健谈和爱交朋友,同时非常欢迎生信、AI多模态领域高手加入团队合作储备人才。

昇腾计算产业是基于昇腾系列(HUAWEI Ascend)处理器和基础软件构建的全栈 AI计算基础设施、行业应用及服务,https://devpress.csdn.net/organization/setting/general/146749包括昇腾系列处理器、系列硬件、CANN、AI计算框架、应用使能、开发工具链、管理运维工具、行业应用及服务等全产业链
更多推荐

 6
6 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)